
The Black Tortoise (Xuan Wu) is a Chinese mythological symbol of reproduction, adapted here to depict the mammalian life cycle (ring) maintained through mating (snake and tortoise), with the red germ cell at the center vital for transmitting genetic information from one generation to the next.
CELL of June 4th, published ‘The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells’ from BIOPIC’s Fuchou Tang’s and Jie Qiao’s groups as its cover story. In this study, the transcriptome as well as DNA methylome of human PGCs from multiple developmental stages were analyzed at single-cell and single-base resolutions, respectively. The researchers found that human PGCs showed unique features very different from mouse PGCs.
Human primordial germ cells (PGCs) are the embryonic founder cells of the mature gametes- the oocytes and sperms. The mature gametes are vital for transmitting both genetic and epigenetic information faithfully from one generation to the next and for maintaining the continuation of a species through fertilization. It is therefore very critical to understand the crucial epigenetic processes of this most fascinating lineage in vivo.
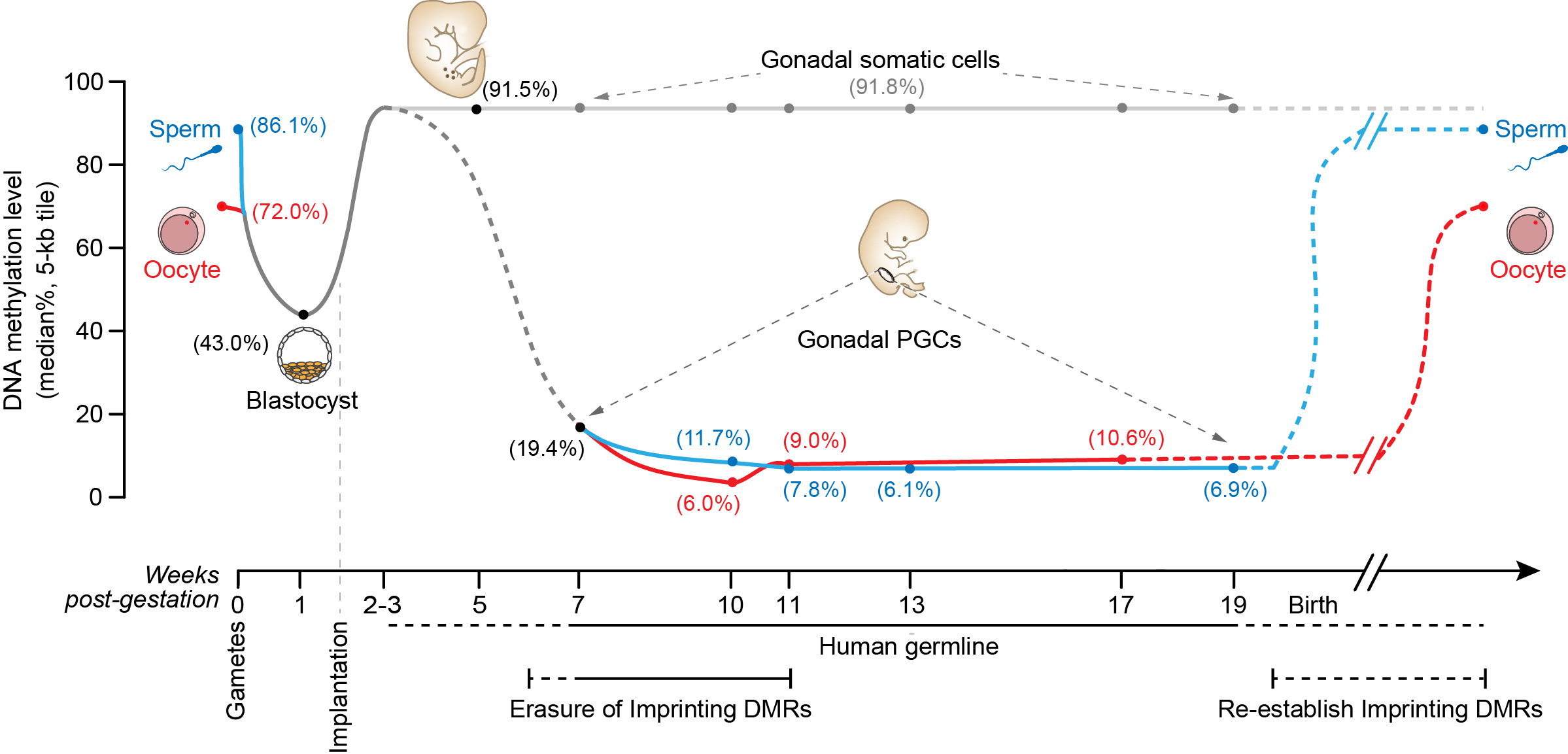
DNA methylation dynamics of human sperm, oocyte, blastocysts, post-implantation embryos, primordial germ cells and gonadal somatic cells.
DNA methylation is one of the most important epigenetic modifications which is vital for embryonic development, stem cell differentiation, cancer and other biological processes. It is heritable without changing the DNA sequences, and can be affected by the external environments. During the past decades, researchers have used mice as one of the model organisms to study the complex regulatory networks between gene transcription and DNA methylation, and have understood the development and epigenetic reprogramming of mouse PGCs well. Two waves of DNA methylation erasure and re-establishment are known during mouse embryonic development: a first wave of global demethylation during preimplantation embryo and a more dramatic demethylation process during PGC development in which germline-specific genes and imprinted genes are thoroughly demethylated. However, the mechanistic understanding of gene expression pattern and epigenetic reprogramming in the human PGCs is still largely unknown due to the limited materials available for analysis.
Recently, the research team led by Prof. Tang at BIOPIC, Peking University has developed a highly sensitive high-throughput sequencing technology used for profiling DNA methylation both at single-base and single-cell resolution. They firstly used this method to profile the DNA methylation landscape of human preimplantation embryos, and found a genome-wide DNA demethylation after fertilization. The DNA methylation level is reduced from 86% (median level) in sperm to 43% in the blastocysts, but the imprinting control region is accurately maintained during this first wave of DNA demethylation. Furthermore, combined with a whole genome bisulfite sequencing (WGBS) strategy, they found that there was a global re-methylation process shortly after implantation and the DNA methylation level reached to 92%. This work has been published in Nature in July, 2014 and provided a invaluable resource for systematically dissecting the gene expression network and epigenetic regulation during human early embryonic development.
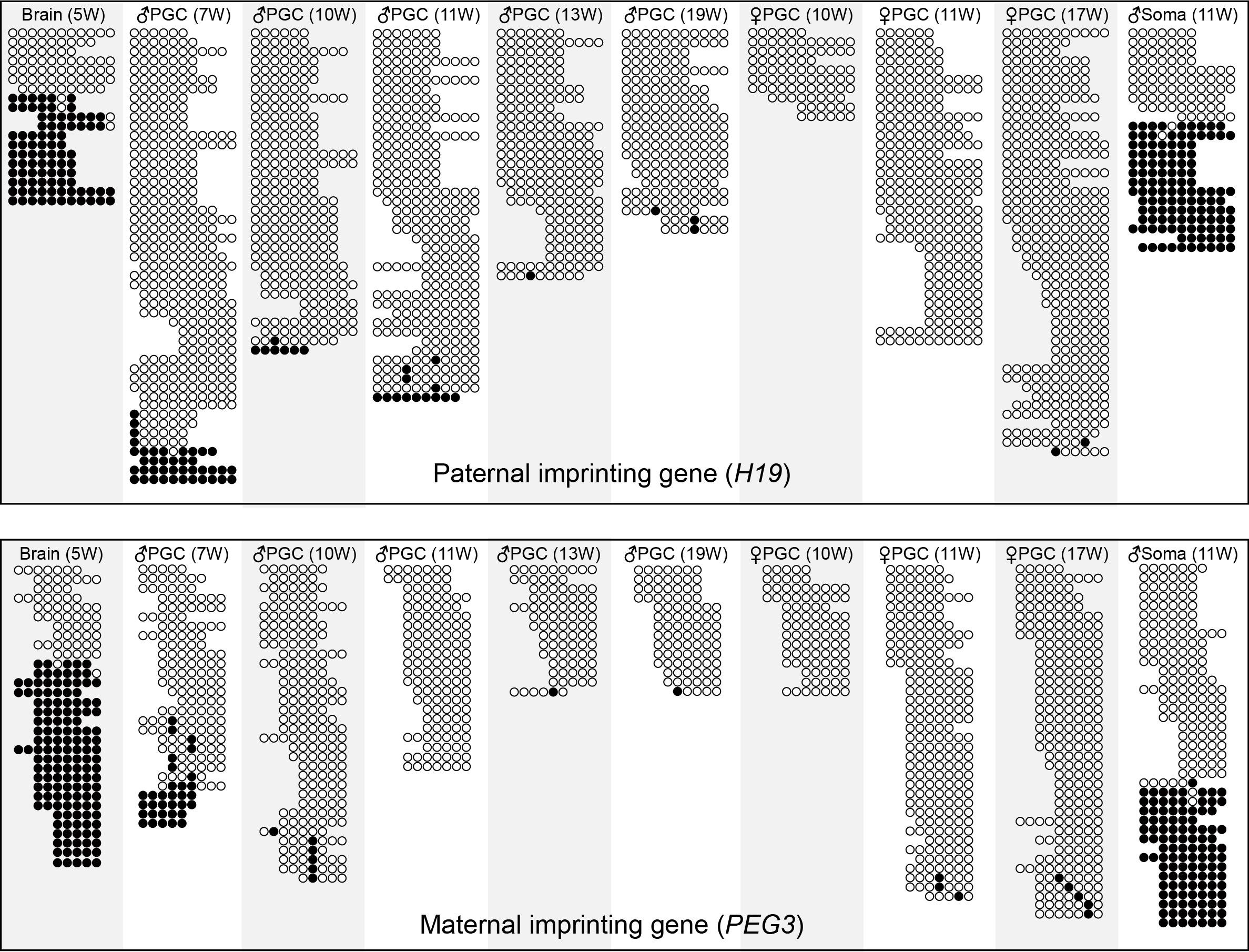
The DNA methylation graphs of paternal imprinting gene H19, maternal imprinting gene PEG3 across different developmental stages. The open white and filled black circles indicate the unmethylated and methylated CpG sites, respectively. And each row in the graphs originated from a single read from the WGBS datasets. Only the reads covered more than 5 consecutive CpG sites were plotted out.
Now, the research team has studied the second round of DNA methylation reprogramming and comprehensively analyzed the relationship between gene expression and DNA methylation in human PGCs. The global DNA methylation level in most tissues and organs remain stable after implantation, but the PGCs which are responsible for transmitting the genetic information to the next generation will undergo a large-scale DNA methylation erasure and re-establishment process. The team used fluorescence-activated cell sorting (FACS) method to enrich for the human PGCs from different developmental stages, then comprehensively analyzed the key features of transcriptome (single-cell RNA-Seq), DNA methylome (PBAT & TAB-Seq) and histone modification (immunofluorescence staining) of human PGCs.
Firstly, Similar to mouse PGCs, human PGCs in early developmental stages express pluripotency genes such as OCT4, NANOG and REX1. However, human PGCs do not express SOX2, instead they express SOX15 and SOX17. Human PGCs also express germline-specific marker genes, such as KIT, TNAP, AP2γ and NANOS3. Different from the mitotic PGCs, there is a strong gene expression heterogeneity of the meiotic PGCs. The PGCs in meiosis exhibit a strong heterogeneity of gene expression among individual cells in the same embryo, probably because they are non-synchronized when entering meiosis arrest. Secondly, one of the two X chromosomes will be randomly inactivated after implantation in female embryos to maintain the same dose of gene expression level from X chromosome in both sexes. However, the genes on the X chromosome exhibit bi-allelic expression in each individual female PGC cells from the migrating stage onward, indicating that the inactivated X chromosome is already reactivated in female PGCs. Thirdly, there is a global DNA demethylation process during the development of human PGCs. The DNA methylation level is deduced from 92% in post-implantation embryos to about 7% in PGCs at 10-11 weeks after gestation. As far as we know, this is the lowest DNA methylation level in any type of normal human cells, indicating a unique feature of DNA methylome of human PGCs. Fourthly, although almost all of the functionally important genomic elements are essentially free of any methylation during the development of human PGCs, some repeat elements, especial the evolutionarily younger and more active ones (such as ALRs (37%), L1 (23%), Alu (12%), and ERVK (30%)) still possess high levels of residual DNA methylation. This observation indicates a basis for potential trans-generation inheritance of epigenetic memory. Lastly, PGCs maintain relatively stable global RNA expression patterns and constitutive heterochromatin status when DNA methylation is globally decreased by more than 10-fold, which indicates other key component of epigenetic regulation, in particular histone modifications probably playing an important role in this process.
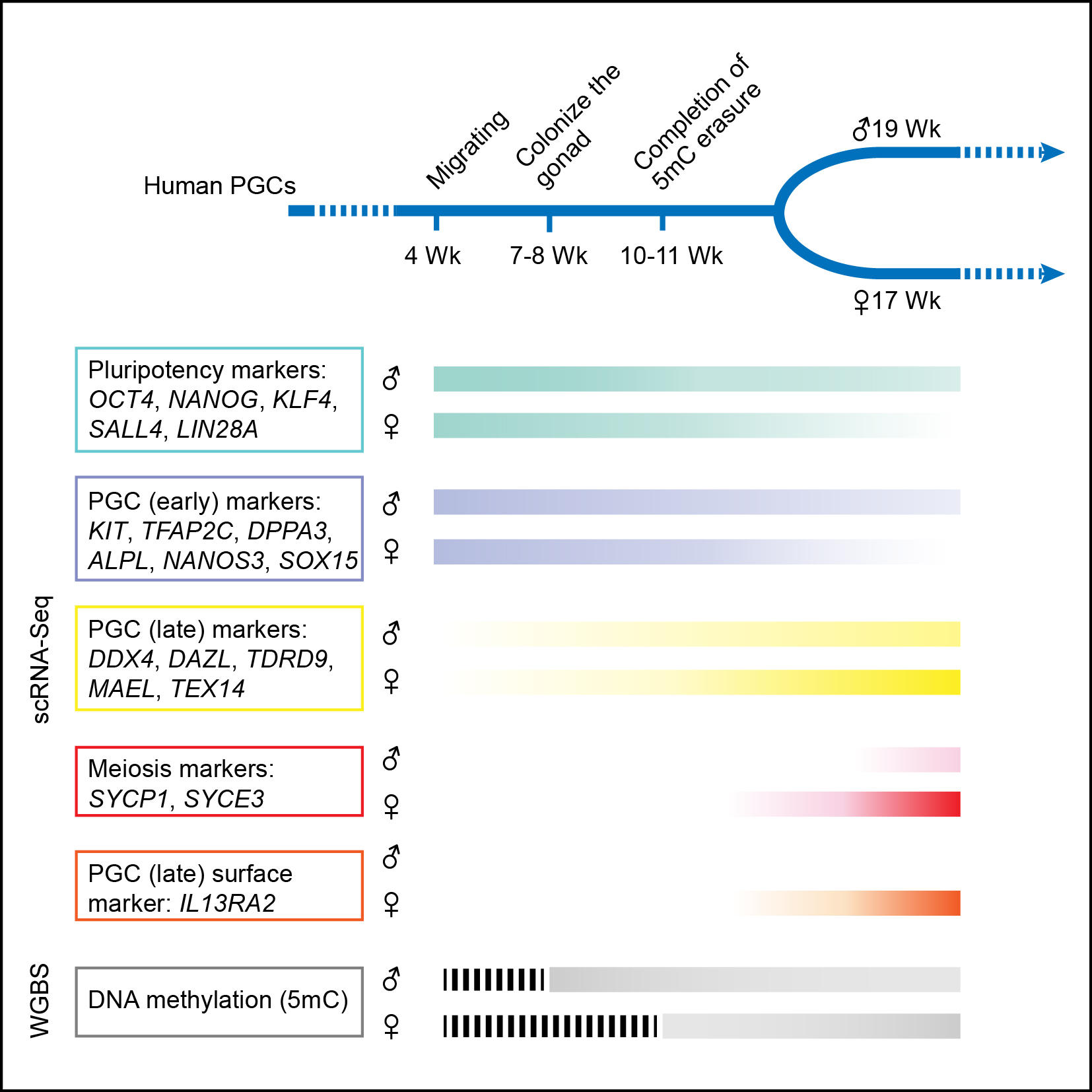
Sketch of the expression patterns of different set of genes and DNA methylation levels during human PGC development.
This is the first time that the transcriptome of human PGCs were comprehensively analyzed at single-cells and single-base resolution, which improves the understanding of development mechanism and epigenetic reprogramming of human PGCs. This work provides basis for exploring the establishment of totipotency in human early embryos, trans-generation inheritance of DNA methylation and generation of the functional gametes in vitro for future studies. And this work also has potential impact on evaluating the safety of assisted reproductive technology, clinical assessment of the epigenetic related diseases and germ cell abnormalities.
Dr. Fan Guo, Dr. Liying Yan, Hongshan Guo and Lin Li are the co-first authors of this paper. Prof. Fuchou Tang at BIOPIC of Peking University and Prof. Jie Qiao at Third Hospital of Peking University are the co-correspondence authors. Prof. Yi Qin Gao’s group at BIOPIC contributed to the bioinformatics analysis of this work. This work was supported by NSF China, the National Basic Research Program of China, the Beijing Municipal Science and Technology Commission, Peking University for translation research and Peking-Tsinghua Center for Life Sciences.
Fan Guo*, Liying Yan*, Hongshan Guo*, Lin Li*, Boqiang Hu, Yangyu Zhao, Jun Yong, Yuqiong Hu, Xiaoye Wang, Yuan Wei, Wei Wang, Rong Li, Jie Yan, Xu Zhi, Yan Zhang, Hongyan Jin, Wenxin Zhang, Yu Hou, Ping Zhu, Jingyun Li, Ling Zhang, Sirui Liu, Yixin Ren, Xiaohui Zhu, Lu Wen, Yi Qin Gao, Fuchou Tang & Jie Qiao. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell 161, 1437–1452. (2015) (*co-first author)
Hongshan Guo*, Ping Zhu*, Liying Yan*, Rong Li*, Boqiang Hu, Ying Lian, Jie Yan, Xiulian Ren, Shengli Lin, Junsheng Li, Xiaohu Jin, Xiaodan Shi, Ping Liu, Xiaoye Wang, Wei Wang, Yuan Wei, Xianlong Li, Fan Guo, Xinglong Wu, Xiaoying Fan, Jun Yong, Lu Wen, Sunney X. Xie, Fuchou Tang & Jie Qiao. The DNA methylation landscape of human early embryos. Nature 511, 606–610. (2014) (*co-first author)